
Report on Assembly Standards
Version 7.0 - January 2026
To accompany the standards, the EBP provides Sequencing and Assembly Recommendations.
The standards in this version have changed from those in the original EBP paper [1] and previous versions (July 2019, July 2020, December 2020, March 2021, May 2023, June 2024), reflecting progress in sequencing and assembly technology. This is a living document, which we aim to revise at least annually (see Appendix C).
Revision 7, October 2025, with input from the wider assembly community.
Agreed by the EBP Sequencing and Assembly Standards committee on 31st Jan 2026.
1. Quantitative assembly standards:
The EBP sequencing and assembly standards committee proposes different assembly standards for three groups of organisms. The three groups are:
(1) Eukaryotic species for which sufficient DNA and tissue is available.
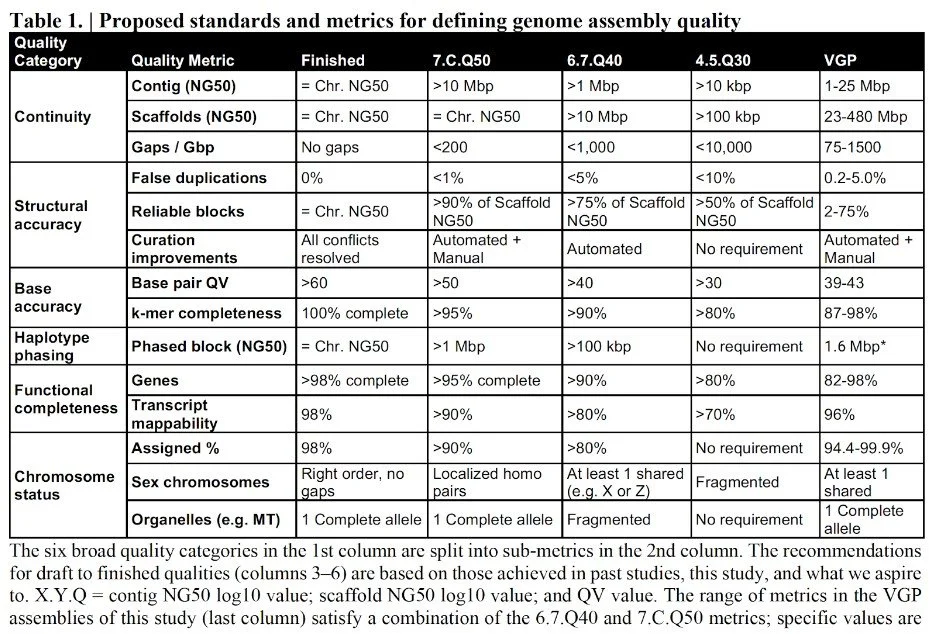
Current long read and long-range (Hi-C) methods require input DNA masses of 0.5 to 10 µg. For most larger species this yield is readily achievable from a single individual. For these species we propose a minimum reference standard of 6.C.Q40, i.e. megabase N50 contig continuity and chromosomal scale N50 scaffolding, with less than 1/10,000 error rate (see table from the VGP flagship assembly paper [2] in Appendix A for notation and further information). For species with chromosome N50 smaller than a megabase this will be C.C.Q40.
Alongside the contiguity and error rate goals we propose the following additional criteria from the table:
< 5% false duplications
> 90% kmer completeness
> 90% sequence assigned to candidate chromosomal sequences
> 90% single copy conserved genes (e.g. BUSCO) complete and single copy (*)
> 90% transcripts from the same organism mappable
Links to standard tools for measuring these metrics are provided in Appendix B below.While we believe that these are achievable goals for most, perhaps ultimately all, species from which large enough, high quality samples can be obtained, we recognise that for many reasons (e.g. sample quality, very large genomes, polyploidy, cost expediency) they may not be met in the first instance. Interim references that do not meet the standard can be very useful and should be submitted to INSDC and valued. However, there should be a continuing EBP goal to revisit them and bring them up to the target standard, as that becomes practical.
(*) The BUSCO requirement is a useful target, but needs to be interpreted in context. It can be relaxed for other reasons than quality of the assembly, first the completeness may be below 90% for taxa that are strongly divergent from those for which the closest BUSCO set was established; second for polyploids or incompletely rediploidized species for which many BUSCO genes may be present in more than one copy, so marked as duplicate by BUSCO. Third, diploid species with large genomes that show no artificial duplications but a considerable fraction of the repeats containing paralogs that increase the % of duplicated BUSCOs.
Recent developments have made yet another approach possible for this organism group: the generation of complete telomere-to-telomere (T2T) assemblies These are usually based on high quality long reads combined with ultralong reads, require significant resources and are therefore not suggested to replace the above mentioned quality standard yet. Reaching T2T quality is defined as the presence of all telomere sequences (where applicable), the absence of sequence gaps and a QV greater than 60 (i.e. scoring C.C.Q60).
(2) Species with limited DNA or material per individual.
For many species the maximum theoretical yield from one individual is less than 100 ng DNA, and this precludes processing through standard library construction and sequencing methodologies. New long range whole genome amplification technologies (also known as Ultra Low Input or ULI) allow data generation sufficient for assembly from as little as 10 ng of DNA. For such species (where total DNA mass is between 100 and 10 ng per individual) it was proposed in May 2023 that a standard of 5.C.Q40 is regularly achievable, i.e. similar to the larger sample target but with contig N50 relaxed to >100kb to reflect amplification dropout.
Advances in sequencing of individual meiofauna (where total DNA mass is <1 ng), following protocols such such as PiMmS and phibeta29 replicase whole genome amplification (WGA), now allow generation of assemblies with a 10e5 contig N50. These assemblies can then be scaffolded, sometimes to chromosomal contiguity, with Hi-C data derived through low-input methods from a pool of individuals. We suggest a minimum of 5.6.Q40 for such species, but encourage achieving of 5.C.Q40 where possible.
(3) Unculturable single cell eukaryotes.
For these we propose a metagenomics-like standard to be determined based on experience in the prokaryotic community. This is still outstanding and – pending the anticipated developments in the coming year (2026) – will be subject of the next update of the standards document.
2. Additional requirements:
Our experience has shown that currently, all (combinations of) automated processes generate assemblies with a variety of remaining errors, some of which are relatively easy to address and should be corrected before submission [3]. We therefore propose that a set of quality control criteria are required to be met including:
a) separation of sequence of the target species from contaminants and other organisms such as symbionts, members of the gut microbiome, pathogens and parasites (collectively “cobionts”)
b) explicit identification of a primary (haploid or pseudo-haploid) assembly, with additional sequence in a secondary bin that may contain either full alternate haplotypes or a set of haplotypic/other sequences from the individual. See below for further discussion of submission of diploid assemblies
c) separation and explicit identification of organellar genomes
d) only A,C,G,T and N bases and sequences should not begin or end with Ns
We would like the majority of chromosome ends to contain telomeric repeat sequence, however this is currently not observed consistently.
We also encourage:
e) identification of discordances between raw data and resulting assembly to locate and remove structural errors (misjoins, missed joins and false duplications);
f) identification and naming of chromosomes, esp. sex chromosomes, where possible (see below);
g) reconciliation with the known karyotype where it exists and this is possible.
h) identification of [within individual] PAR (pseudoautosomal region) annotations of sex chromosomes in the form of annotation and/or softmasking of the W/Y PAR, while recognising that this may be difficult in practice for some many species.
i) assembly and submission of all cobionts present at sufficient abundance in the sequenced sample;
j) > 50% telomere/telomere-like structure completeness, recognising that not all species possess canonical telomeres;
k) a read coverage profile that is uniform and coherent with the type of assembly (primary + alts, hap1/hap2, …) submitted; and
l) if possible, orient the chromosome sequence with the p arm first and keep it consistent between haplotypes, so all haplotypes are in the same orientation.
Identification and naming of chromosomal-scale scaffolds can be achieved by consulting Hi-C 2D maps and comparison to existing karyotyping and linkage maps. If chromosome naming already exists for a given species, the naming and orientation should be reflected in the new assembly. If no previous naming exists, we recommend to name chromosomes by size, taking into account scaffolds that can be assigned to belong to a certain chromosome, but could not be unambiguously placed (unlocalised scaffolds). An alternative that is applicable in some cases is to name chromosomes after those in a closely related species with established nomenclature; we only recommend this if there are no major interchromosomal rearrangements identified between the species, i.e. all chromosomes are in one-to-one correspondence, but may have within chromosome rearrangements. The method for assignment of chromosome names should be clearly stated in the submission metadata. In case a previous assembly did orient the chromosomes incorrectly (see (l)), reorient them in the correct way (p before q) in the current assembly.
3. INSDC project structure and nomenclature:
For a reference genome to count towards the EBP goals it must be submitted to the INSDC (GenBank/EMBL/DDBJ) Genomes Division for open access use by the scientific community.
When this submission is made the assembly is associated with a BioProject object, which can be part of a hierarchy by being assigned to an “umbrella” BioProject. We suggest the structure in Figure 1 below, with a data project for the raw data and one per assembly, an umbrella project for the target species (note that under this there may be assemblies of separate symbiont or cobiont species), and then above an umbrella BioProject corresponding to the overall project. Please also link your top level BioProject to the EBP BioProject object, whose identifier is PRJNA533106 (https://www.ncbi.nlm.nih.gov/bioproject/533106). If you are an EBP affiliate project and need PRJNA533106 to link down to your project please contact Erich Jarvis, (ejarvis@rockefeller.edu). If the assembly is also contributing to other larger scale efforts such as e.g. GIGA, ERGA or VGP then you can also link your assembly to their umbrella BioProjects.
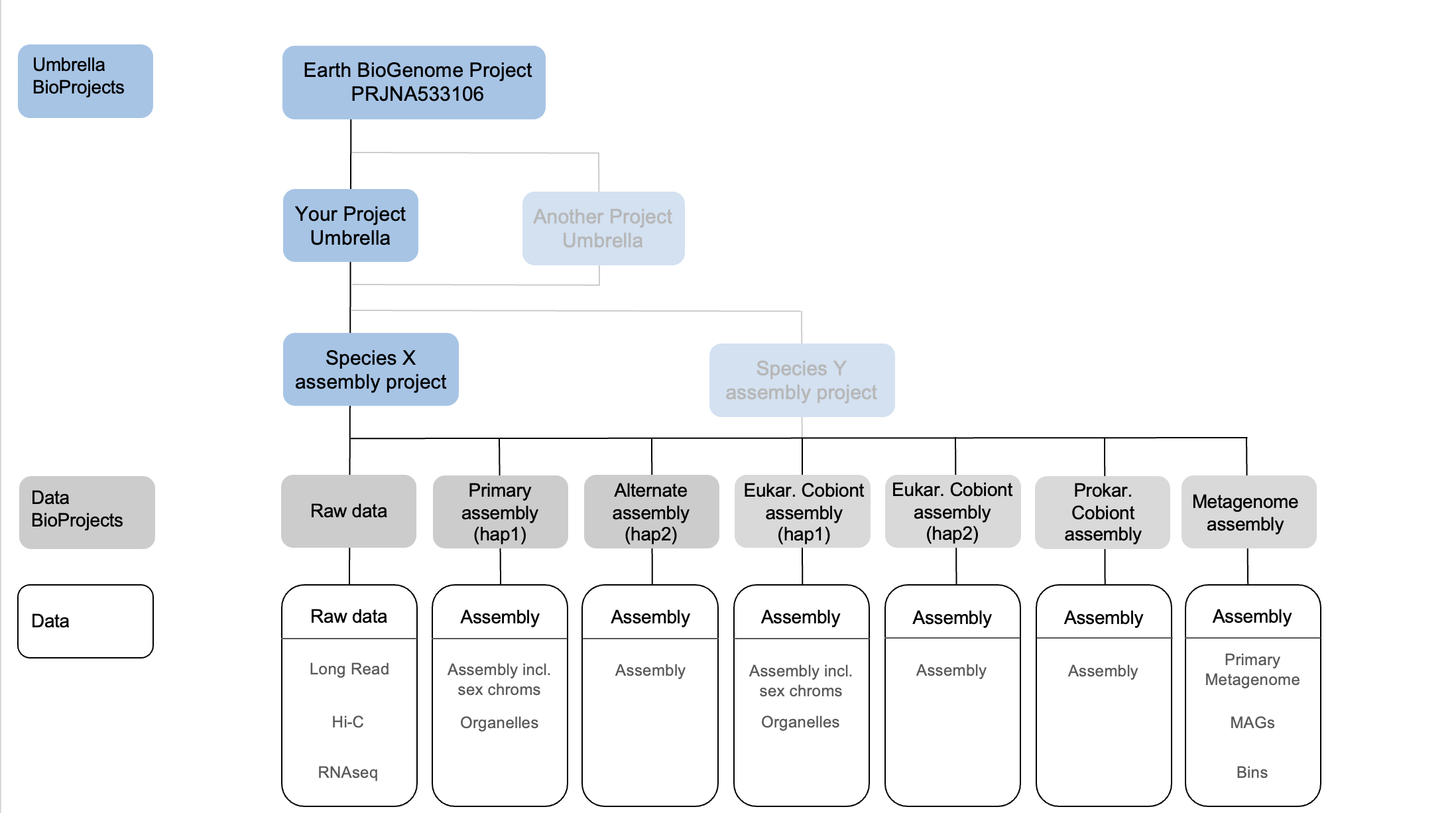
Figure 1: Overview of the INSDC BioProject, assembly and raw data submission structure. Add haplotype assembly BioProjects and data as required in case of polyploid eukaryotes. Equally, add as required for multiple prokaryotic assemblies, e.g. Wolbachias. Eukaryotic cobionts will require their own tolid and BioProject. Umbrella projects can be linked and stacked to reflect their relationships. If several individuals of the same species are assembled and released by the same project, their data BioProjects can be added to the species umbrella BioProject.
As well as connecting to a BioProject, an assembly needs to be assigned to a “txid” entry in the NCBI Taxonomy database. Although txid identifiers can be created for informal taxa such as Maylandia sp. “pearly”, we would prefer EBP genomes to be associated with taxonomically valid species names, and urge EBP-affiliated projects to work with appropriate taxonomists to identify samples to a species and where necessary to establish the species name in the standard manner in the literature. Txid identifiers at levels below species (e.g. subspecies or strains) will be tracked by EBP at the species level.
Furthermore, in addition to the numerical identifiers generated for assemblies by the public databases we request projects to adopt the tolid (for Tree of Life ID) standard short nomenclature for samples and assemblies, as used by the VGP and Darwin Tree of Life project. This takes the form <clade><gen><spec><ind>.<assembly> e.g. ilAlcRepa1.1 for the first assembly of insect lepidoptera Alcis repandata individual 1. Unique species designations have been generated to cover all ~485,000 species with data in INSDC or found in Britain and Ireland with relatively few clashes that were resolved with a simple process; other species can be added on request. From summer 2024, tolids can also be assigned to subspecies, variants or other taxonomic levels below “species”. The assigned tolids will reflect the parent species to preserve the ability of EBP species level accounting. In case a pool of specimen was sequenced, a unique tolid can be assigned to the specific pool. A server to view and assign unique individual identifiers for samples is available at https://id.tol.sanger.ac.uk/, where you can also find details on the two letter prefix assignments which assigns all 26 letters to a top level partition of the tree of life. For queries please contact tolid-help@sanger.ac.uk. See Blaxter et al. [5] for an introduction to tolids, txid and notes on obtaining txid for currently unrepresented species
Finally, it is mandatory for EBP assemblies to submit to INSDC the primary raw data used to build the assembly along with the primary assemblies. For ENA submissions, the raw data needs to be referred to in the RUN_REF fields of the manifest of the assembly submission, as indicated in the instructions at https://ena-docs.readthedocs.io/en/latest/submit/assembly/genome.html#manifest-files. For Genbank submissions, instructions can be found here: https://www.ncbi.nlm.nih.gov/sra/docs/submit/. Submission of assemblies and raw reads with the above recommended Bioproject umbrella structure will ensure linking and discoverability.
4. diploid assemblies:
History: This section was added to version 5 following a discussion in the EBP/VGP assembly call on 5 May 2023.
It is now possible to separate out homologous chromosomes during assembly to provide two essentially complete sets of chromosomes. There are two standard approaches to this. The first uses sequence data from the parents to phase the chromosomes, resulting in a maternal haploid genome and a paternal haploid genome, and a fully parentally phased assembly. We recommend that these are called <tolid>.mat and <tolid>.pat and to provide an additional synthetic genome including both X and Y for annotation purposes. The second uses long range phasing data from Hi-C or (ultra)long reads to phase individual chromosomes, resulting potentially in fully phased individual chromosomes, or if not then ones with few phase switches, but no phase coherence across chromosomes. In the latter case we recommend that the resulting haploid genome sets are called <tolid>.hap1 and <tolid>.hap2. For polyploids, e.g. tetraploids, the corresponding nomenclature would be <tolid>.mat1, .mat2, .pat1, .pat2 or .hap1, .hap2, .hap3, .hap4. We are suggesting that reasonable efforts are being made where possible to assort the relevant subgenomes into submitted haplotypes. This can e.g. be based on kmer similarity between repeats/TEs.
Despite submitting two full haploid genomes that are effectively equivalent in quality, the wider community still for many purposes uses a single “primary” haploid reference genome sequence. By default this will be .hap1 if phased with long range data. For trio-phased assemblies the primary assembly will need to be designated (or a synthetic reference constructed that contains both heterogametic sex chromosomes).
For species that have heterogametic karyotypes (X0, XY, ZW, and variants thereof) sequencing of the heterogametic sex (male or female as relevant) is strongly recommended.. Because it is desired that the primary reference contains as complete a representation as possible of the species’ genome in haploid form, we recommend that .hap1 should contain the entirety of all known sex chromosomes, along with metadata describing the location of any pseudoautosomal regions and/or the non-recombining region (to allow for downstream masking of the recombining sequences, but to prevent loss of information. . An important requirement is that all the material should come from the same individual, and that the sum of the material in all haplotype assemblies should give the complete diploid/polyploid genome. For X0 or Z0 species .hap1 should contain the X or Z chromosome. For U/V species, .hap1 should include the U and V; sequencing one sexed individual (male or female) will likely preclude the identification of the sex chromosomes.
We suggest adding annotations of putative sex chromosomes to the assembly description, whilst we are working with INSDC to possibly implement a nomenclature indicating the unsure nature of these identification that can be directly applied as chromosome name.
In some cases, where an individual of the homogametic sex has been sequenced, people have added a heterogametic sex chromosome from another individual to make a reference for mapping, e.g. adding a Y from a male when the sequenced individual was an XX female. This is deprecated in EBP primary submissions. We request that instead a synthetic reference for alignment is made using material from two different primary assembly submissions. This also applies to U/V species, where a synthetic reference should contain one haplotype and both the U and the V chromosomes.
The organellar (mitochondrion, plastid etc.) genomes should be included in the primary assembly.
5. Conclusion:
We believe the standards specified above are an essential foundation for high quality genome annotation and analysis supporting the scientific and societal goals of the EBP. However, we note the need for and encourage research to address the following problems and increase the proportion of species reaching this assembly standard: 1. Generating uniform coverage of long read sequences from low input DNA, 2. High quality phased assembly of high heterozygosity and polyploid genomes 3. Better genome sequencing and assembly of mixtures of unculturable single cell eukaryotes.
No specific sequencing recommendations are made, to allow for future technology improvement and change. We urge EBP affiliated projects to share their sequencing recipes and assembly pipelines via publication, protocols.io and github for reuse by others. Multiple long read plus HiC and/or other scaffolding strategies, for example the Vertebrate Genomes Project assembly pipeline (available on Galaxy) or the Tree of Life assembly pipeline, now regularly reach these standards given sufficient material for less than the projected EBP phase 1 cost of $30,000 (1). Indeed we believe that it is now possible to reach these standards at substantially lower cost, of the order of $5,000 in direct costs for a 1 Gb genome, though we note that real costs vary widely across different countries. Because of this rapid change, the committee is optimistic about reducing sequencing complexity and computational costs in the next few years to achieve the estimated direct cost for phases 2 and 3 of the EBP of $800/species for EBP (in 2018 dollars (1)). To monitor this we will continue to revisit the standards on at least an annual basis, reconsidering what is feasible in terms of accuracy, completeness and cost.
1. H. A. Lewin et al., Earth BioGenome Project: Sequencing life for the future of life. Proceedings of the National Academy of Sciences of the United States of America 115, 4325-4333 (2018). https://www.pnas.org/doi/10.1073/pnas.1720115115
2 Rhie et al., Towards complete and error-free genome assemblies of all vertebrate species. Nature 592:737–746 (2021) https://doi.org/10.1038/s41586-021-03451-0
3. Howe K et al., Significantly improving the quality of genome assemblies through curation. GigaScience 10(1) (2021) https://doi.org/10.1093/gigascience/giaa153
4. Blaxter M et al., The Earth BioGenome Project Phase II: illuminating the eukaryotic tree of life. Front Sci 3 (2025) https://doi.org/10.3389/fsci.2025.1514835
5. Blaxter M, Pauperio J, Schoch C, Howe K, Taxonomy Identifiers (TaxId) for Biodiversity Genomics: a guide to getting TaxId for submission of data to public databases (2024) https://doi.org/10.12688/wellcomeopenres.22949.1
Appendix A: Historical table of proposed metrics from 2018
from Rhie et al, bioRxiv https://doi.org/10.1101/2020.05.22.110833
Appendix B: Tools to obtain EBP assembly metrics
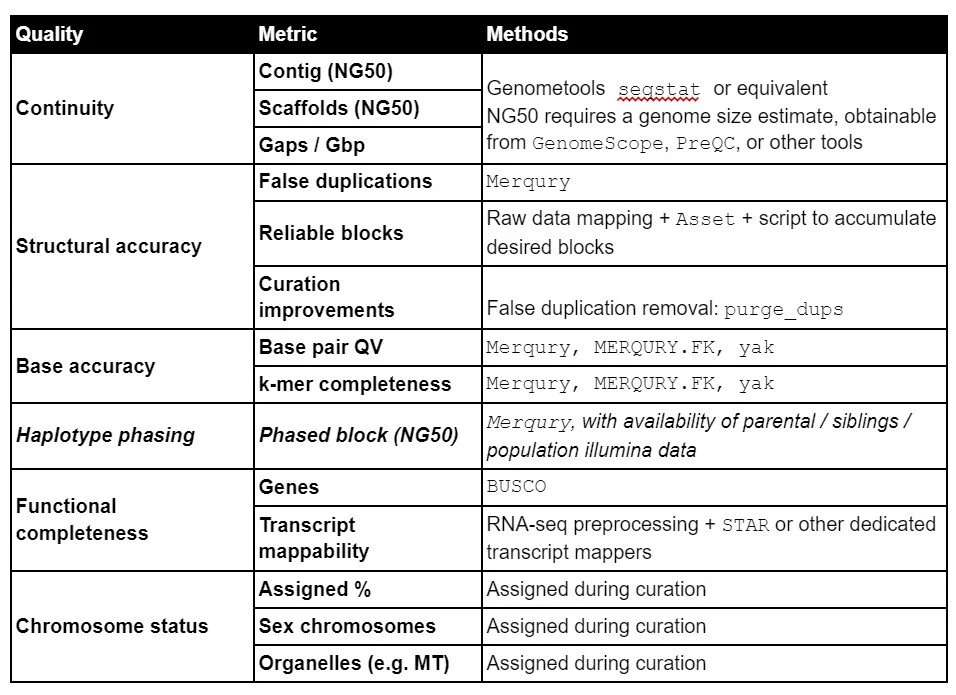
Genometools: http://genometools.org/tools.html
Merqury: https://github.com/marbl/merqury
Purge_dups: https://github.com/dfguan/purge_dups
NB1 Merqury requires a high accuracy shotgun data set (Illumina or PacBio CCS) to extract kmers from. Ideally this will be independent from the one used to build the primary assembly.
NB2 Validation of haplotype phasing accuracy requires genome-wide sequence from a close relative so is not possible in many cases. It should be carried out where possible, but is not required to meet the EBP assembly standard.
Appendix C: Evolution of the EBP sequencing and assembly standards document
Summary of changes from document versions 4 to 7, aided by chatGPT+

ABOUT THE SUBCOMMITTEE
This Report on Assembly Standards was developed by EBP’s Scientific Subcommittee for Sequencing and Assembly.